Research

05 Jun 2020
Characterization of the familial risks of developing atypical hemolytic uremic syndrome (aHUS)
A work now published in the journal Blood by the laboratory of Prof. Santiago Rodríguez de Córdoba in the Centro de Investigaciones Biológicas Margarita Salas and Ciber de Enfermedades Raras has provided robust estimates of the familial risks of developing atypical hemolytic uremic syndrome (aHUS) associated with rare complement mutations.
The group of Prof. Rodríguez de Córdoba, is an international reference for the genetics of aHUS a rare life-threatening disease that in most cases progresses to end-stage kidney disease. At one time, the group used genetics to unravel the pathogenic mechanism that today supports treatment of aHUS patients with eculizumab, a humanized monoclonal antibody that inhibits complement, and that has changed the natural history of the disease.
Nowadays, they routinely perform genetic analyses to reinforce the clinical diagnosis of the disease and to facilitate a personalized medicine in aHUS patients. However, these genetic analyses unravel numerous asymptomatic relatives carrying complement pathogenic mutations, which raises much concern among families and clinicians because of the lack of reliable aHUS penetrance values for these individuals.
Arjona et al. have analyzed data generated from 103 aHUS pedigrees, including almost 500 individuals -remarkable numbers for an ultra-rare disease like aHUS- providing robust estimates of the risk of developing aHUS associated with different loads of rare complement mutations and how these risks are modified by the presence of common aHUS risk polymorphisms.
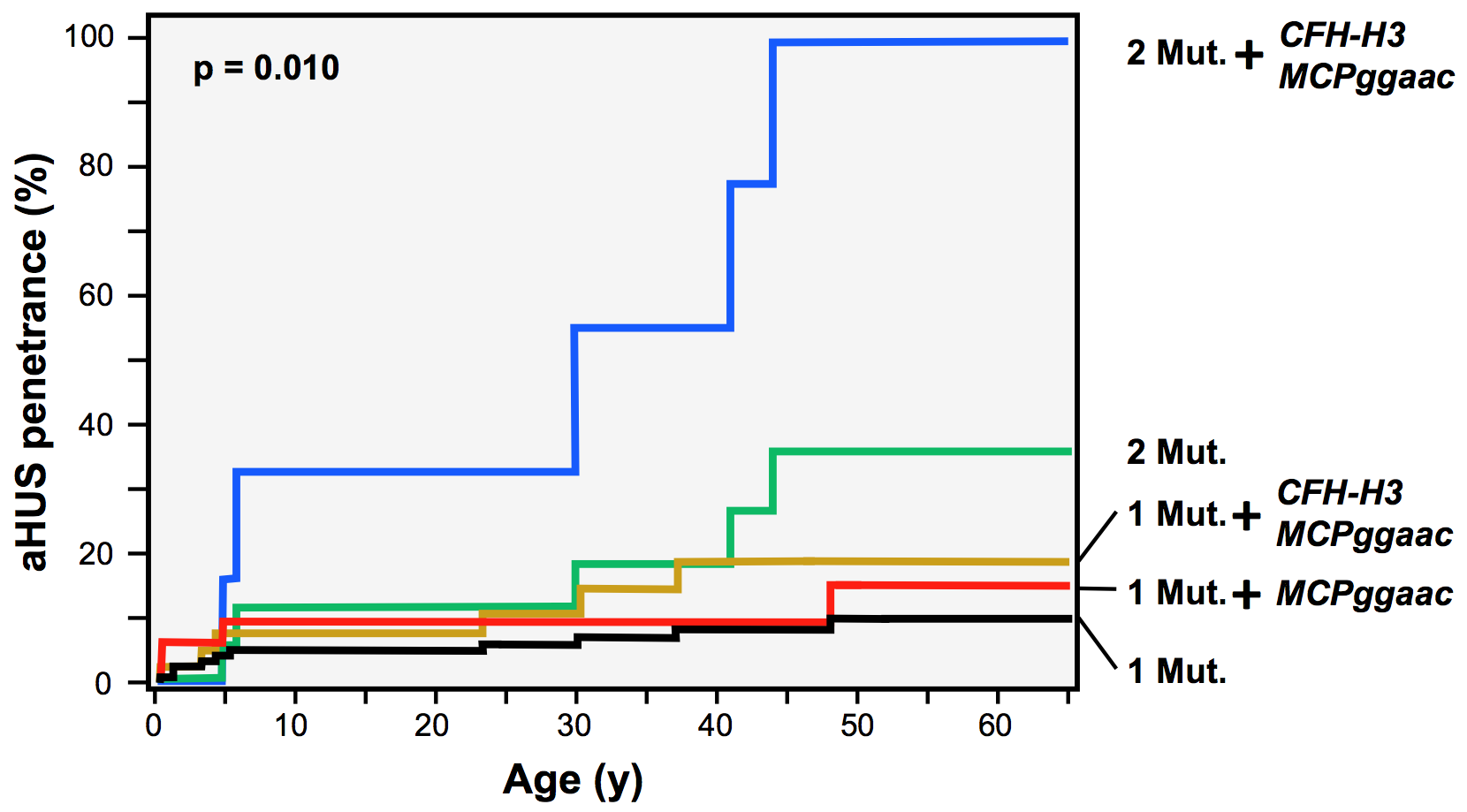
The data provided should be very valuable to clinicians to improve tailored management of aHUS patients and will facilitate genetic counseling to their family members.
This work has been performed in collaboration with the Hospital Puerta de Hierro and the Universidad Complutense de Madrid.
Reference: The familial risk of developing atypical Hemolytic Uremic Syndrome. Emilia Arjona, Ana Huerta, Elena Goicoechea de Jorge, Santiago Rodríguez de Córdoba (2020) Blood. doi.org/10.1182/blood.2020006931
More information:
CIBERER press release: link